Research Areas
Molecular Dynamics of Biomolecules
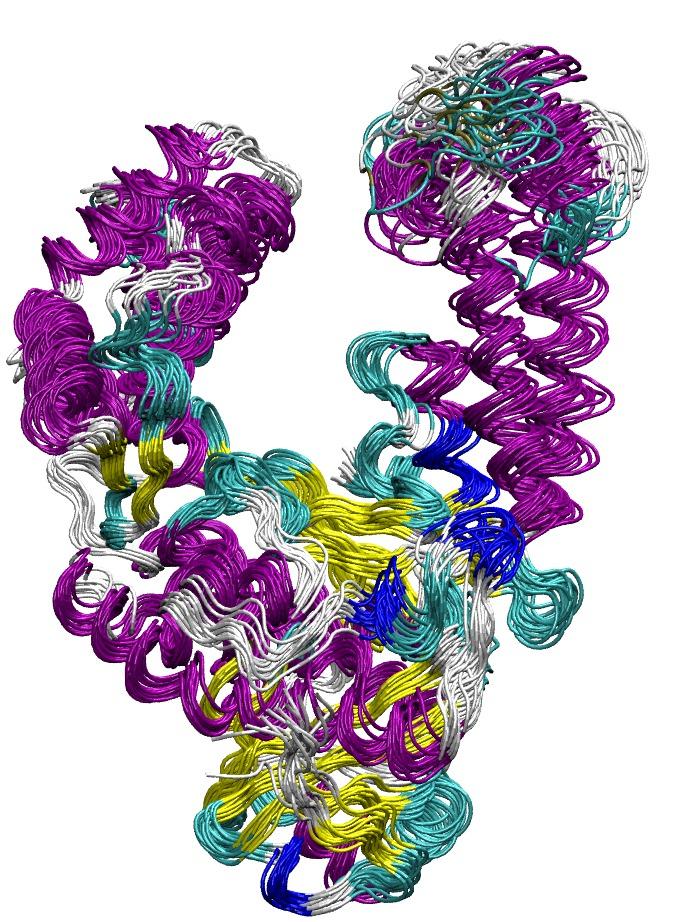
Molecular Dynamics (MD) simulations can be used to follow the dynamics of biomolecules at high spatial (atomistic) and high time (femtosecond) resolution. In order to be functional proteins and nucleic acids need to undergo conformational transitions after being synthesized in a cell to adopt a correctly folded and functionally active form. The active structure still undergoes conformational changes essential for its biological activity. The coupling of function and dynamics is not well understood and often difficult to investigate experimentally. We use the MD technique to study the structure formation process of small proteins and structural motifs in nucleic acids. In addition, MD-based methods are applied to calculate free energy changes associated with structural transitions of proteins and nucleic acids upon binding to partner molecules.
Flexible Docking of Biological Macromolecules
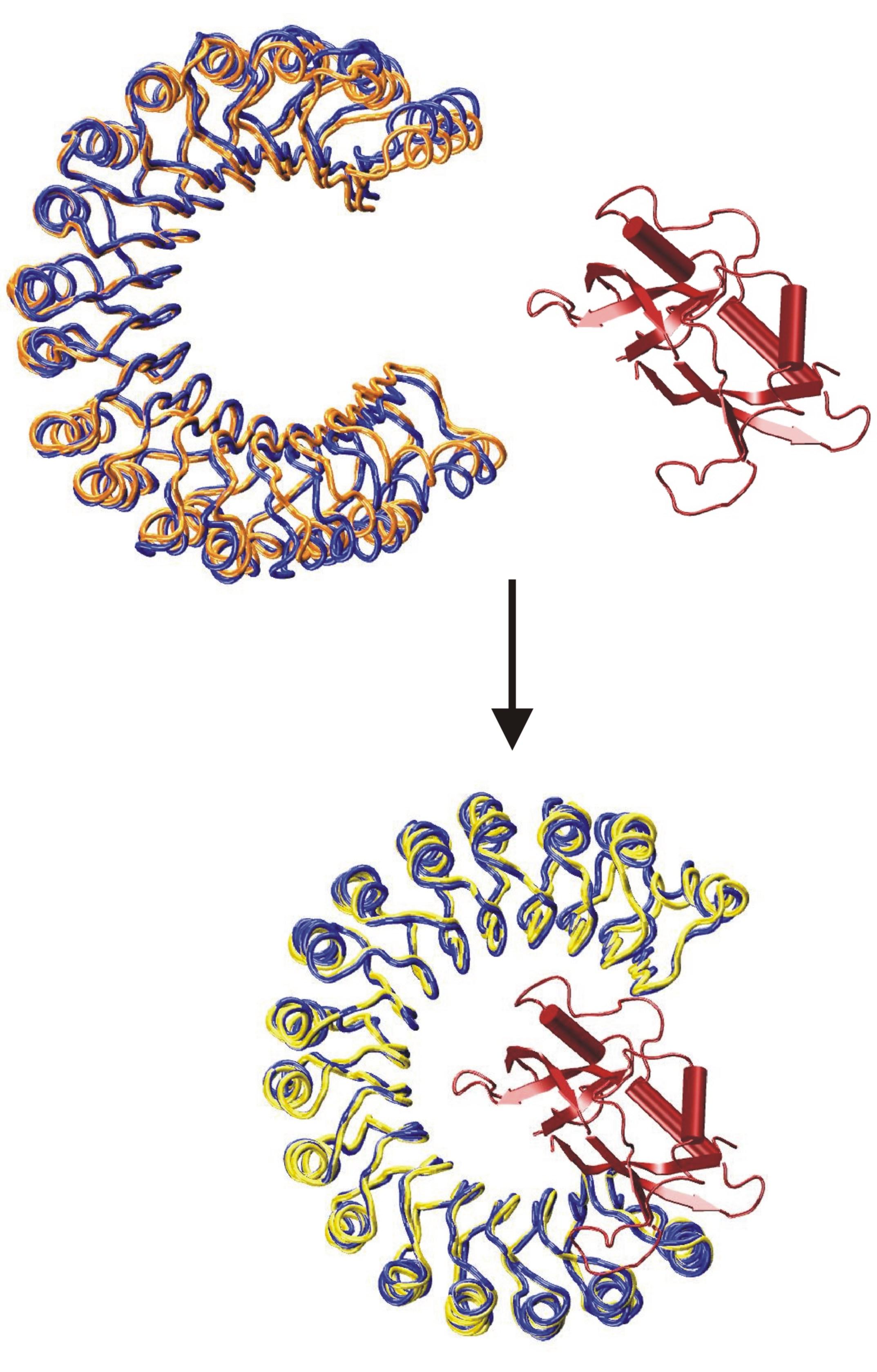
Basically all biological processes involve the interaction and complex formation of biomolecules. A full understanding of these interactions requires the three-dimensional (3D) structure of interacting complexes. Experimental structure determination is only possible for a fraction of all possible complexes and may not be feasible for all low-affinity or transient interactions. Therefore, the realistic prediction of protein-protein complexes and protein-nucleic acid complexes is of great importance. We have developed a powerful docking methodology (ATTRACT) based on a coarse-grained representation of partner molecules. The method allows the efficient inclusion of certain types of conformational changes during the computational docking process. The approach is one of the best performing methods in the blind community-wide docking challenge CAPRI. Main focus of current research is on improving the inclusion of conformational flexibility and the extension to multiple protein-protein interactions. Recently, we have also been able to extend the methodology for docking protein-RNA and protein-DNA complexes.
Prediction of Ligand-Protein Binding
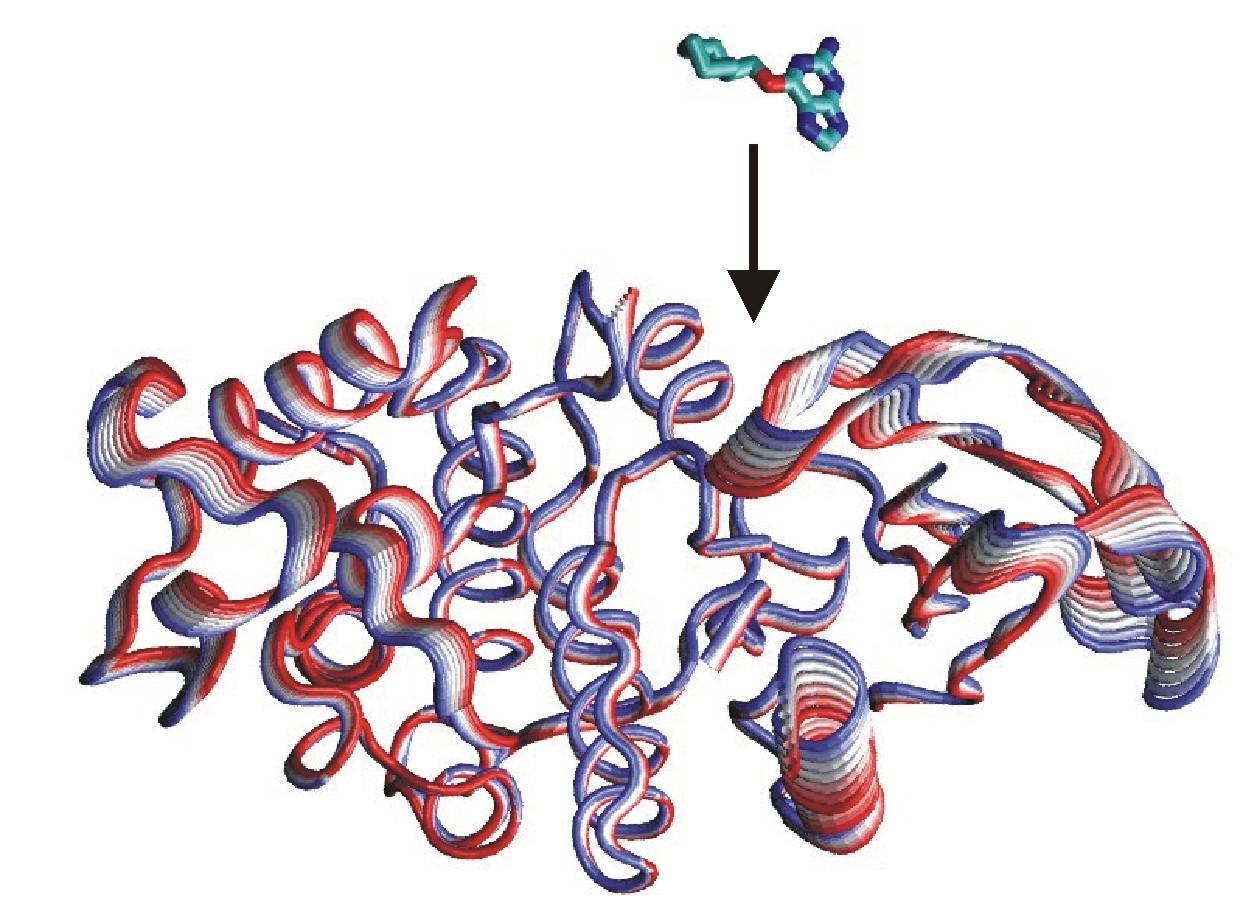
Understanding where and why drug-like ligands bind to specific sites on biological target molecules is of major biophysical and pharmaceutical importance. Our research efforts aim at better understanding the physical basis of affinity and specificity of ligand binding. In addition, we focus on developing improved methods to predict the binding of ligands to protein molecules. Efficient treatment of conformational changes during docking to receptor molecules is a major computational challenge. A new docking methodology has been developed that includes ligand flexibility and both global backbone flexibility and side chain flexibility of the protein receptor. In a recent effort it was also possible to combine global collective receptor flexibility with an efficient grid-based representation of the receptor molecules. This approach shows promising results on docking drug-like compounds to several kinase systems and the conformationally flexible HIV protease.
Development of Advanced Sampling MD-Techniques
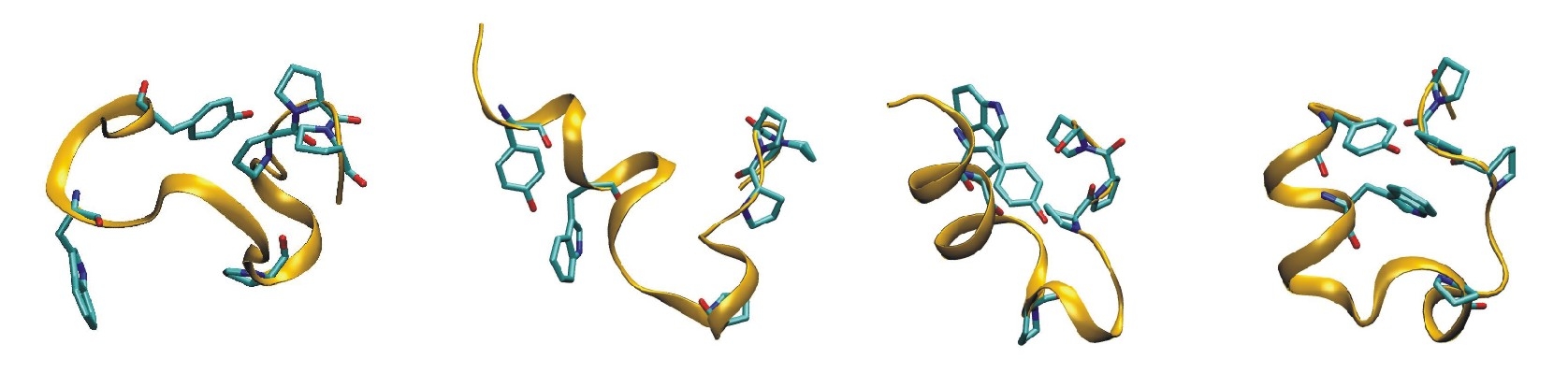
During replica exchange molecular dynamics (REMD) simulations several replicas of a system are simulated at different temperatures in parallel allowing for exchange between replicas at frequent intervals. This technique can improve sampling of conformational space. A drawback of the standard temperature REMD is the rapid increase of the replica number with increasing system size to cover a desired temperature range. We develop Hamiltonian-REMD methods specifically designed to enhance the sampling of protein and nucleic acid conformations by applying various levels of a backbone biasing potential for each replica run.