Recent Publications
Efficient Refinement and Free Energy Scoring of Predicted Protein–Protein Complexes Using Replica Exchange with Repulsive Scaling
T. Siebenmorgen, M. Zacharias
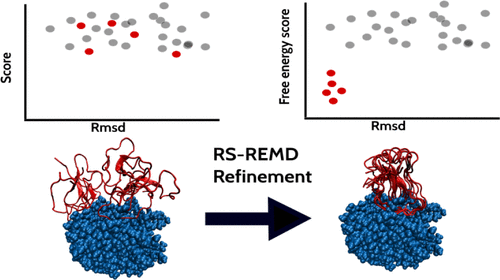
Accurate prediction and evaluation of protein–protein complex structures are of major importance to understand the cellular interactome. Typically, putative complexes are predicted based on docking methods, and simple force field or knowledge-based scoring functions are applied to evaluate single complex structures. We have extended a replica-exchange-based scheme employing different levels of a repulsive biasing between partners in each replica simulation (RS-REMD) to simultaneously refine and score protein–protein complexes. The bias acts specifically on the intermolecular interactions based on an increase in effective pairwise van der Waals radii (repulsive scaling (RS)-REMD) without affecting interactions within each protein or with the solvent. The method provides a free energy score that correlates quite well with experimental binding free energies on a set of 36 complexes with correlation coefficients of 0.77 and 0.55 in explicit and implicit solvent simulations, respectively. For a large set of docked decoy complexes, significant improvement of docked complexes was found in many cases with the starting structure in the vicinity (within 20 Å) of the native complex. In the majority of cases (14 out of 20 in explicit solvent), near native docking solutions were identified as the best scoring complexes. The approach is computational demanding but may offer a route for refinement and realistic ranking of predicted protein–protein docking geometries.
Rapid in silico Design of Potential Cyclic Peptide Binders Targeting Protein-Protein Interfaces
B. L. Santini, M. Zacharias
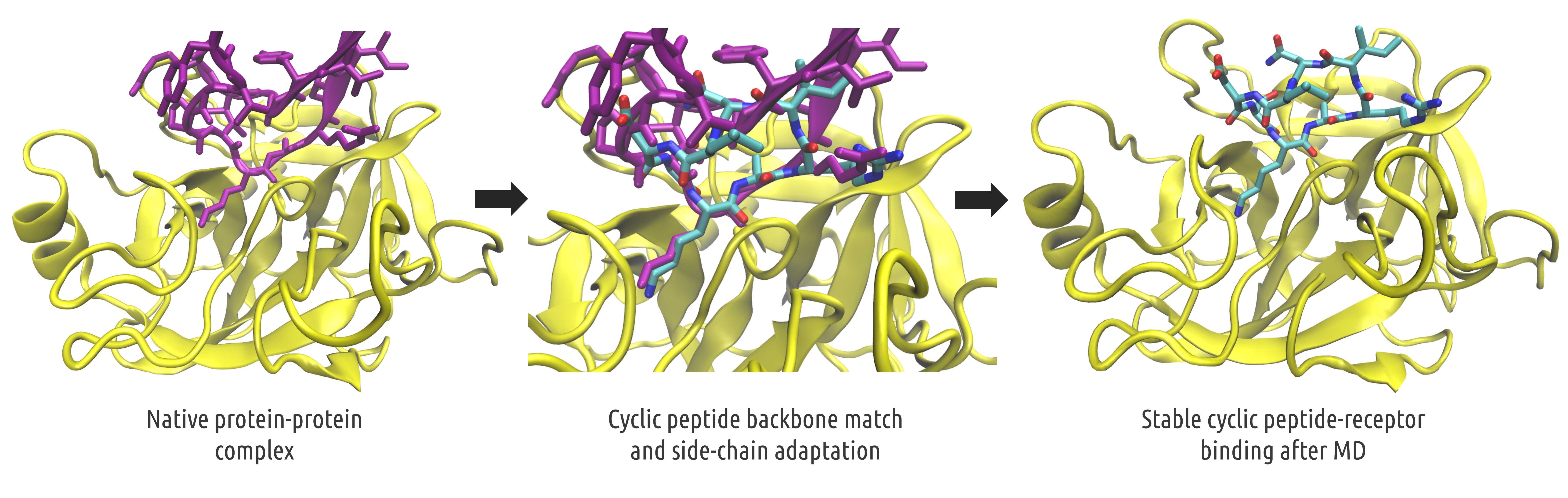
Rational design of specific inhibitors of protein-protein interactions is desirable for drug design to control cellular signal transduction but also for studying protein-protein interaction networks. We have developed cPEPmatch, a rapid computational approach to rationally design cyclic peptides that potentially bind at desired regions of the interface of protein-protein complexes. The methodology is based on comparing the protein backbone structure of short peptide segments (epitopes) at the protein-protein interface with a collection of cyclic peptide backbone structures. A cyclic peptide that matches the backbone structure of the segment is used as a template for a binder by adapting the amino acid side chains to the side chains found in the target complex.
How Mutations Perturb γ-Secretase Active Site Studied by Free Energy Simulations
S.-Y. Chen, M. Zacharias
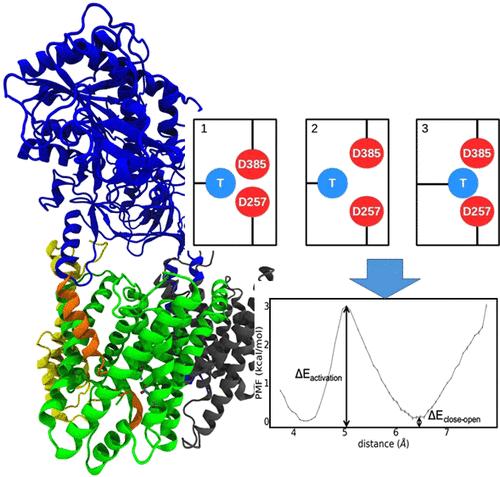
γ-Secretase is involved in processing of the amyloid precursor protein (APP) and generation of short Aβ peptides that may play a key role in neurodegenerative diseases such as Alzheimer’s disease (AD). Several mutations in γ-secretase influence its activity, resulting in early AD onset (Familial AD or FAD mutations). The molecular details of how mutations, not located close to the active site, can affect enzyme activity is not understood. In molecular dynamics simulations of γ-secretase in the absence of substrate (apo), we identified two active site conformational states characterized by a direct contact between catalytic Asp residues (closed state) and an open water-bridged state. In the presence of substrate, only conformations compatible with the open active site geometry are accessible. Systematic free energy simulations on wild type and FAD mutations indicate a free energy difference between closed and open states that is significantly modulated by FAD mutations and correlates with the corresponding experimental activity. For mutations with reduced activity, an increased penalty for open-state transitions was found. Only for two mutations located at the active site a direct perturbation of the open-state geometry was observed that could directly explain the drop of enzyme activity. The simulations suggest that modulation of the closed/open equilibrium and perturbation of the open (active) catalytic geometry are possible mechanisms of how FAD mutations affect γ-secretase activity. The results also offer an explanation for the experimental finding that FAD mutations, although not located at the interface to the substrate, mainly destabilize the enzyme–substrate complex.
How methyl sugar interactions determine DNA structure and flexibility
K. Liebl, M. Zacharias
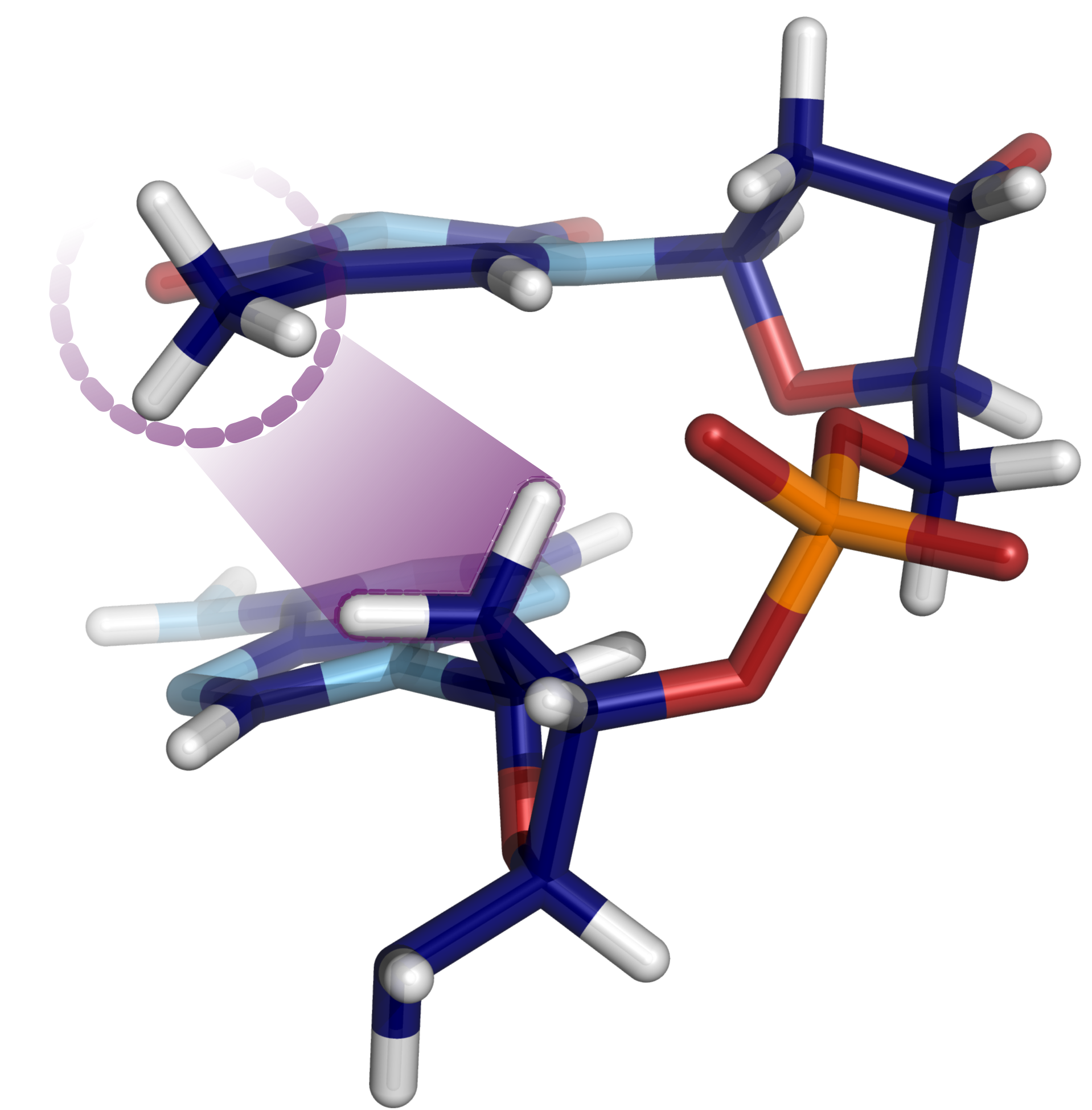
The sequence dependent structure and flexibility of the DNA double helix is of key importance for gene expression and DNA packing and it can be modulated by DNA modifications. The presence of a C5′-methyl group in thymine or the frequent C5′-methylated-cytosine affects the DNA fine structure, however, the underlying mechanism and steric origins have remained largely unexplained. Employing Molecular Dynamics free energy simulations that allow switching on or off interactions with the methyl groups in several DNA sequences, we systematically identified the physical origin of the coupling between methyl groups and DNA backbone fine structure. Whereas methyl-solvent and methyl–nucleobase interactions were found to be of minor importance, the methyl group interaction with the 5′ neighboring sugar was identified as main cause for influencing the population of backbone substates. The sterical methyl sugar clash prevents the formation of unconventional stabilizing hydrogen bonds between nucleobase and backbone. The technique was also used to study the contribution of methyl groups to DNA flexibility and served to explain why the presence of methyl sugar clashes in thymine and methyl-cytosine can result in an overall local increase of DNA flexibility.
Atomic Resolution Insight into Sac7d Protein Binding to DNA and Associated Global Changes by Molecular Dynamics Simulations
M. Zacharias
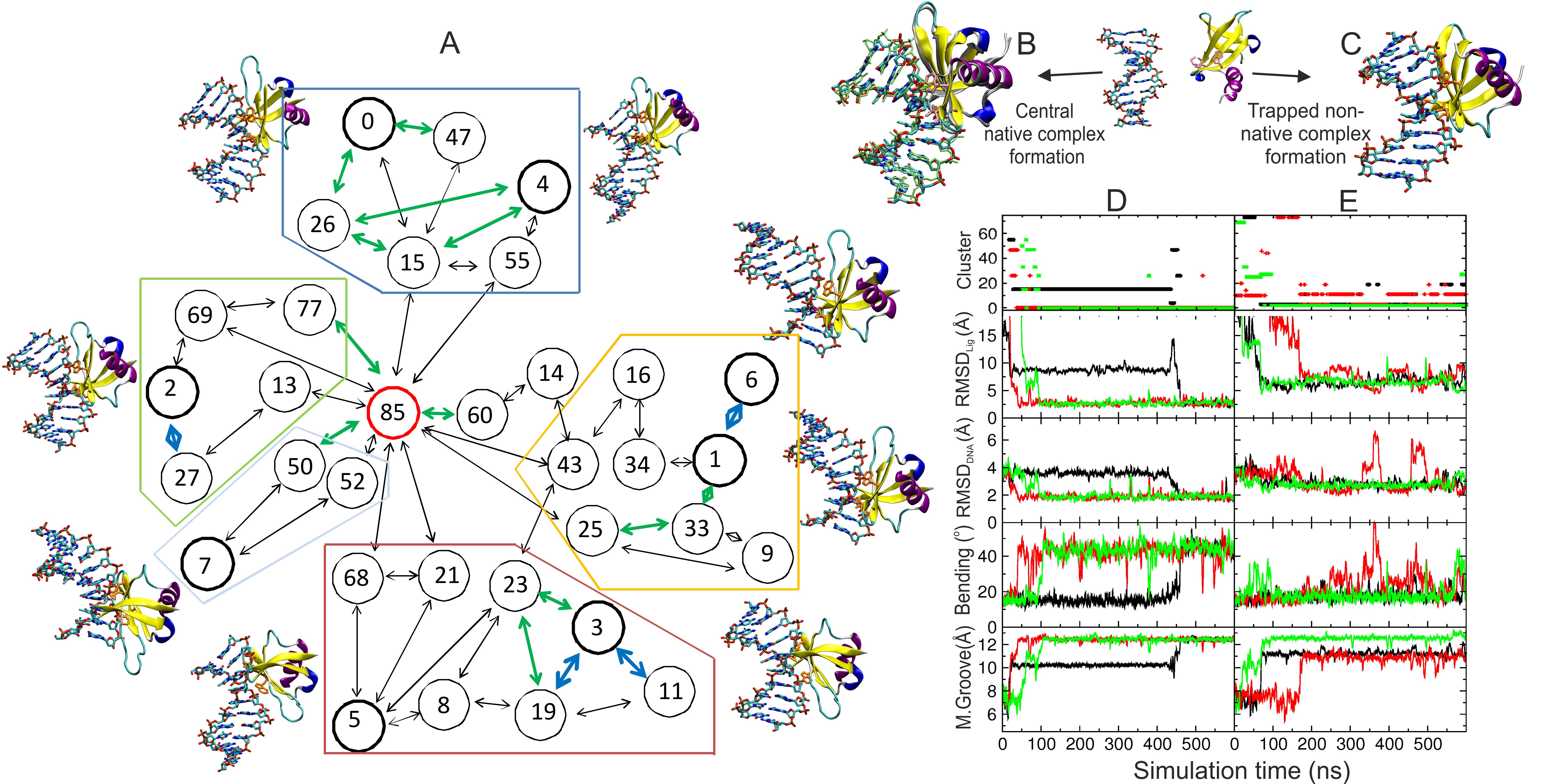
Sac7d is a small, thermostable protein that induces large helical deformations in DNA upon association. Starting from multiple initial placements of the unbound Sac7d structure relative to a B-DNA oligonucleotide, molecular dynamics (MD) simulations were employed to directly follow several successful binding events at atomic resolution that resulted in structures in close agreement with the native complex geometry. The final native complex formed rapidly within tenths of nanoseconds and included simultaneous large-scale kinking, groove opening, twisting, and intercalation in the target DNA. The simulations indicate that the complex formation process involves initial non-native contacts that helped in reaching the final bound state, with residues intercalated at the center of the kinked DNA. It was also possible to identify several long-lived trapped intermediate states of the binding process and to follow sliding processes of Sac7d along the DNA minor groove.
Evaluation of Predicted Protein-Protein Complexes by Binding Free Energy Simulations
T. Siebenmorgen, M. Zacharias
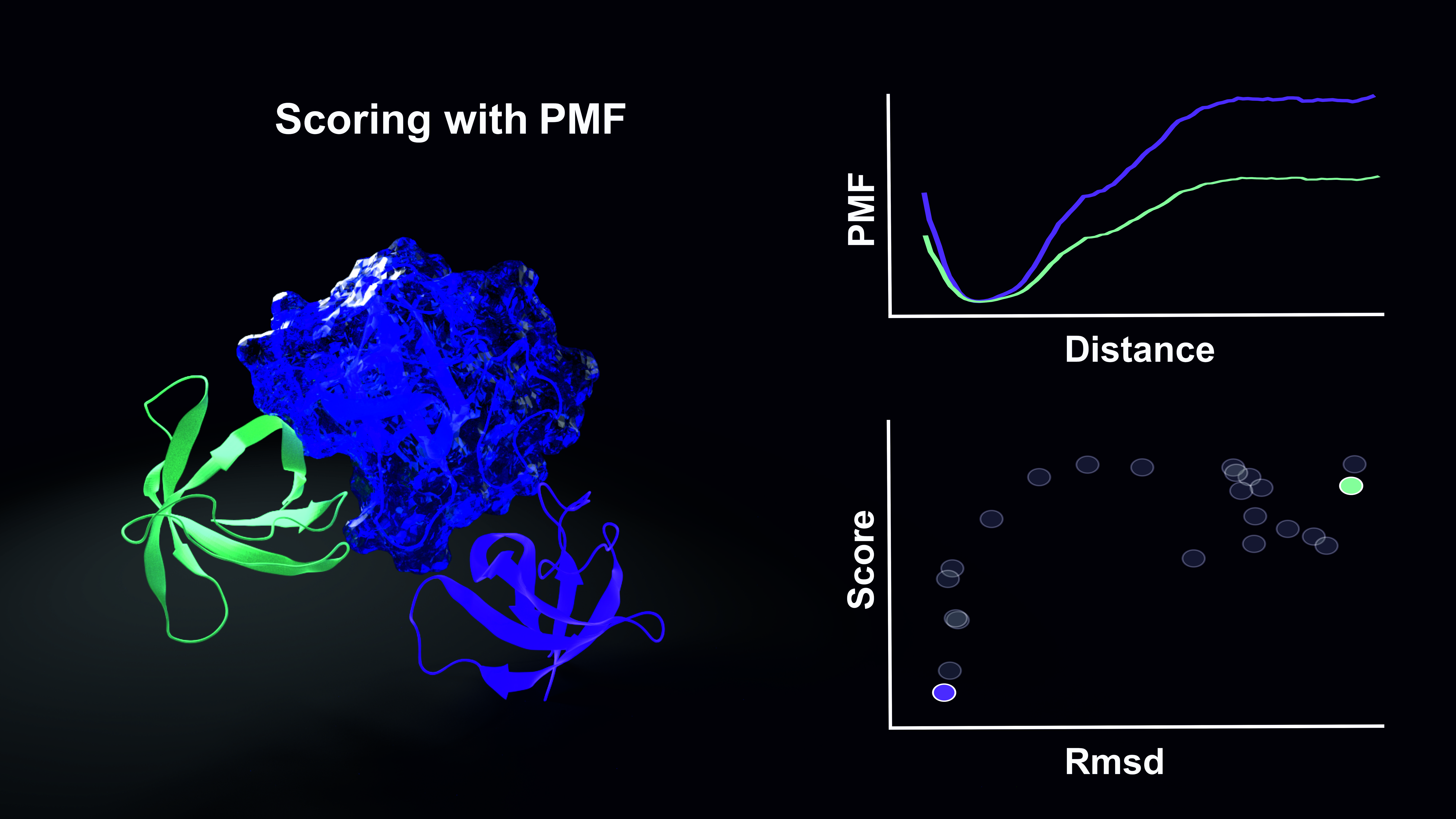
The accurate prediction of protein-protein complex geometries is of major importance to ultimately model the complete interactome of interacting proteins in a cell. A major bottleneck is the realistic free energy evaluation of predicted docked structures. Typically, simple scoring functions applied to single-complex structures are employed that neglect conformational entropy and often solvent effects completely. The binding free energy of a predicted protein-protein complex can, however, be calculated using umbrella sampling (US) along a predefined dissociation/association coordinate of a complex. We employed atomistic US-molecular dynamics simulations including appropriate conformational and axial restraints and an implicit generalized Born solvent model to calculate binding free energies of a large set of docked decoys for 20 different complexes. Free energies associated with the restraints were calculated separately. In principle, the approach includes all energetic and entropic contributions to the binding process. The evaluation of docked complexes based on binding free energy calculation was in better agreement with experiment compared to a simple scoring based on energy minimization or MD refinement using exactly the same force field description. Even calculated absolute binding free energies of structures close to the native binding geometry showed a reasonable correlation to experiment. However, still for a number of complexes docked decoys of lower free energy than near-native geometries were found indicating inaccuracies in the force field or the implicit solvent model. Although time consuming the approach may open up a new route for realistic ranking of predicted geometries based on calculated free energy of binding.
Structural Modeling of γ-Secretase Aβ n Complex Formation and Substrate Processing
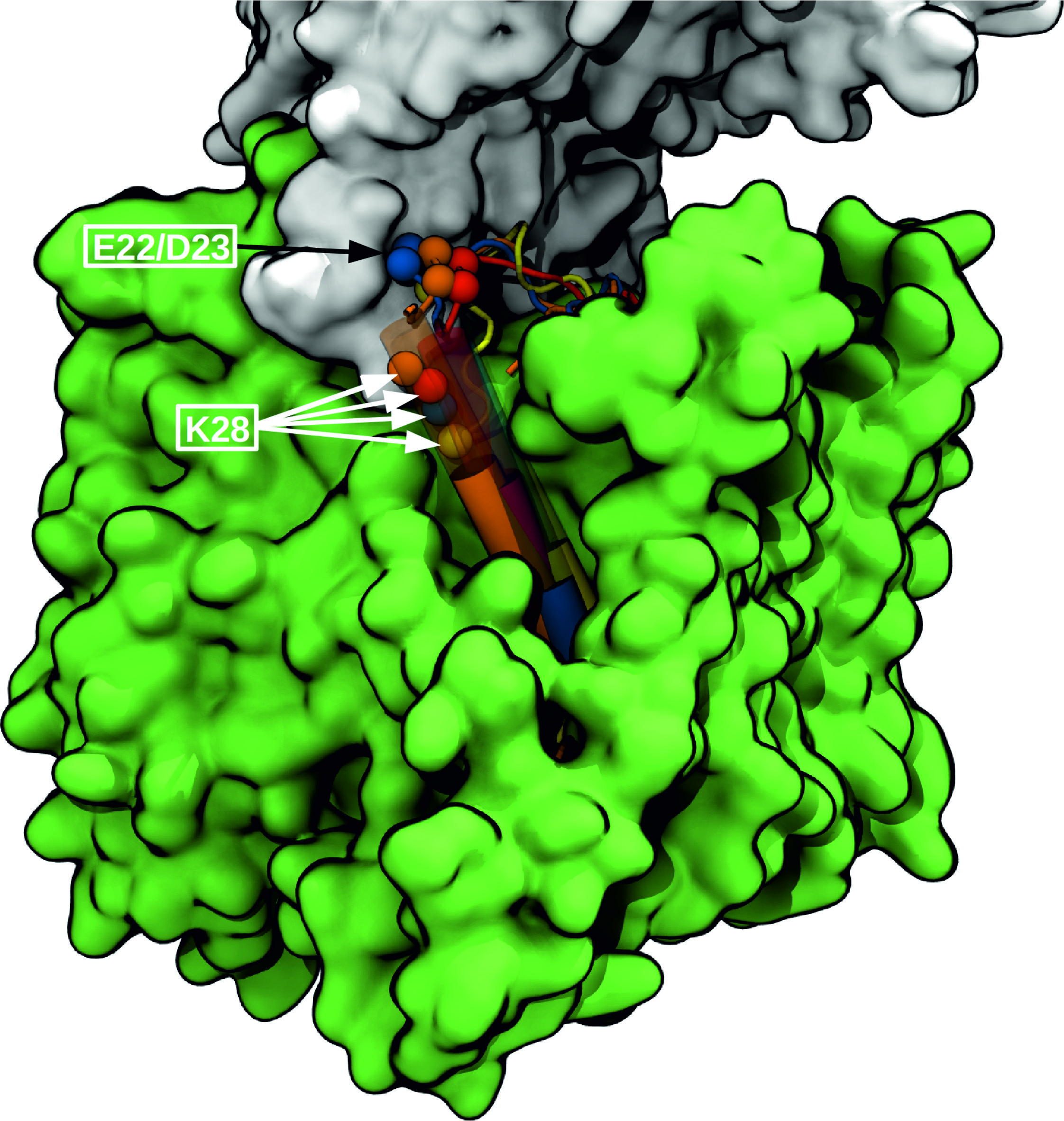
The intramembrane aspartyl protease γ-secretase (GSEC) cleaves single-span transmembrane helices including the C-terminal fragment of the amyloid precursor protein (APP). This substrate is initially cleaved at the ϵ-site followed by successive processing (trimming) events mostly in steps of three amino acids. GSEC is responsible for the formation of N-terminal APP amyloid-β (Aβ) peptides of different length (e.g., Aβ42) that can form aggregates involved in Alzheimer's disease pathogenesis. The molecular mechanism of GSEC-APP substrate recognition is key for understanding how different peptide products are formed and could help in designing APP-selective modulators. Based on the known structure of apo GSEC and the APP-C99 fragment we have generated putative structural models of the initial binding in three different possible modes using extensive molecular dynamics (MD) simulations. The binding mode with the substrate helix located in a cleft between the transmembrane helices 2 and 3 of the presenilin subunit was identified as a most likely binding mode. Based on this arrangement, the processing steps were investigated using restraint MD simulations to pull the scissile bond (for each processing step) into a transition like (cleavable) state. This allowed us to analyze in detail the motions and energetic contributions of participating residues. The structural model agrees qualitatively well with the influence of many mutations in GSEC and C99. It also explains the effects of inhibitors, cross-linking, as well as spectroscopic data on GSEC substrate binding and can serve as working model for the future planning of structural and biochemical studies.