Development of Advanced Sampling MD-Techniques
Biophysical systems are typically complex and corresponding partition functions cannot be calculated analytically. In order to determine free energies, canonical ensembles can be sampled using MD-simulations. Convergence and efficiency of the sampling strongly depend on a convenient choice of integration pathways and sampling methods. A major challenge consists in the treatment of large barriers in the systems' free energy landscapes that are typically not overcome within accessible time scales in standard simulations.
During temperature replica exchange molecular dynamics (T-REMD) simulations, several replicas of a system are sampled at different temperatures. This technique can improve sampling of conformational space, as at higher temperatures free energy barriers can easier be overcome. A drawback of the standard T-REMD is the rapid increase of the replica number with increasing system size to cover a desired temperature range. We develop Hamiltonian-REMD (H-REMD) methods specifically designed to enhance the sampling of protein and nucleic acid conformations by applying various levels of biasing potentials to each replica simulation. By only changing a specific part of the Hamiltonian the sampling can be greatly enhanced while keeping the number of replicas at the minimum. We also combine H-REMD with other free energy calculation methods like for instance Umbrella Sampling (US). In this technique, a system is driven along a reaction coordinate in several umbrella simulations by artificial additional harmonic (umbrella) potentials. Conformational exchanges between these umbrella simulations via H-REMD can significantly contribute to an efficient convergence.
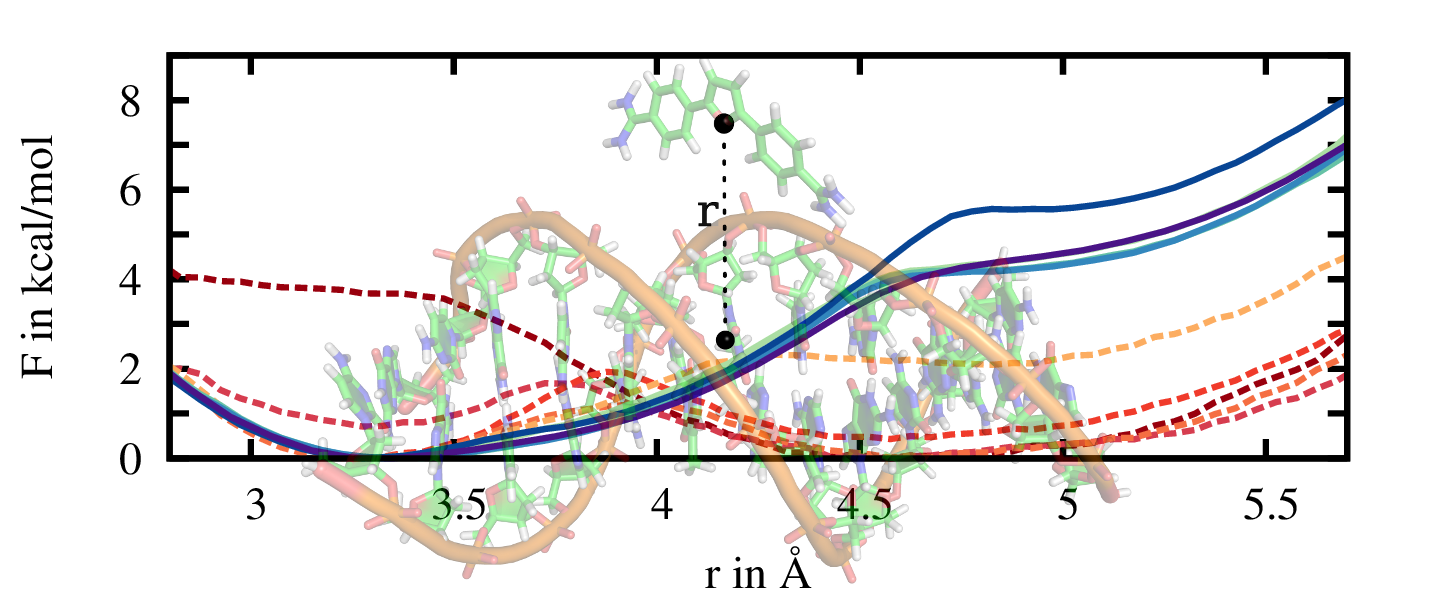
The sampling along specific coordinates via US can be greatly enhanced by smoothening rough free energy landscapes via adaptive biasing potentials (ABPs). We use previously calculated free energy predictions and subtract them iteratively from the system's Hamiltonian by applying the corresponding force. In this way, the system is systematically driven in high free energy regions of interest that would be hardly visited within standard simulations. Ideally, the ABP leads to an equally distributed sampling along the entire reaction coordinate.