Novel Antibacterial Targets
The Challenge:
There is an urgent need for new antibiotic therapies due to a rising population of pathogens that have acquired resistances against multiple antibiotics. Despite their great success in the treatment of infectious diseases, a large fraction of current antibiotics suffer from two major limitations: 1. Classical antibiotics kill bacteria and tragically this is their weak point: bacteria rapidly divide resulting inevitably in spontaneous mutations. If such a mutation eventually prevents killing, the corresponding bacteria will grow in the presence of the drug – termed resistance. 2. Many antibiotics address only a limited number of essential cellular targets including cell wall biosynthesis, DNA replication and protein biosynthesis. Resistance mechanisms against these targets are well established.
Our Goals:
A major research goal of our group is the development of new chemical strategies to fight multiresistant bacterial strains. In order to overcome the limitations of classical antibiotics we follow two major aims: 1. To avoid the classical antibiotic mechanism of killing and selection we exploit chemical approaches that globally disarm bacterial pathogenesis. Bacteria secrete a huge arsenal of toxins – termed virulence factors - that harm the human body. We seek to identify global regulators of virulence and identify small molecules for their selective inhibition. This strategy is attractive as bacteria are not killed and thus have a lower tendency to develop resistance. 2. We search for novel cellular targets in pathogenic bacteria beyond the classical antibiotic mode of action. Targeting different cellular pathways is advantageous as they are devoid of established resistance mechanisms. Thus, chemical compounds that selectively address these targets are likely effective in killing pathogens.
Our Research:
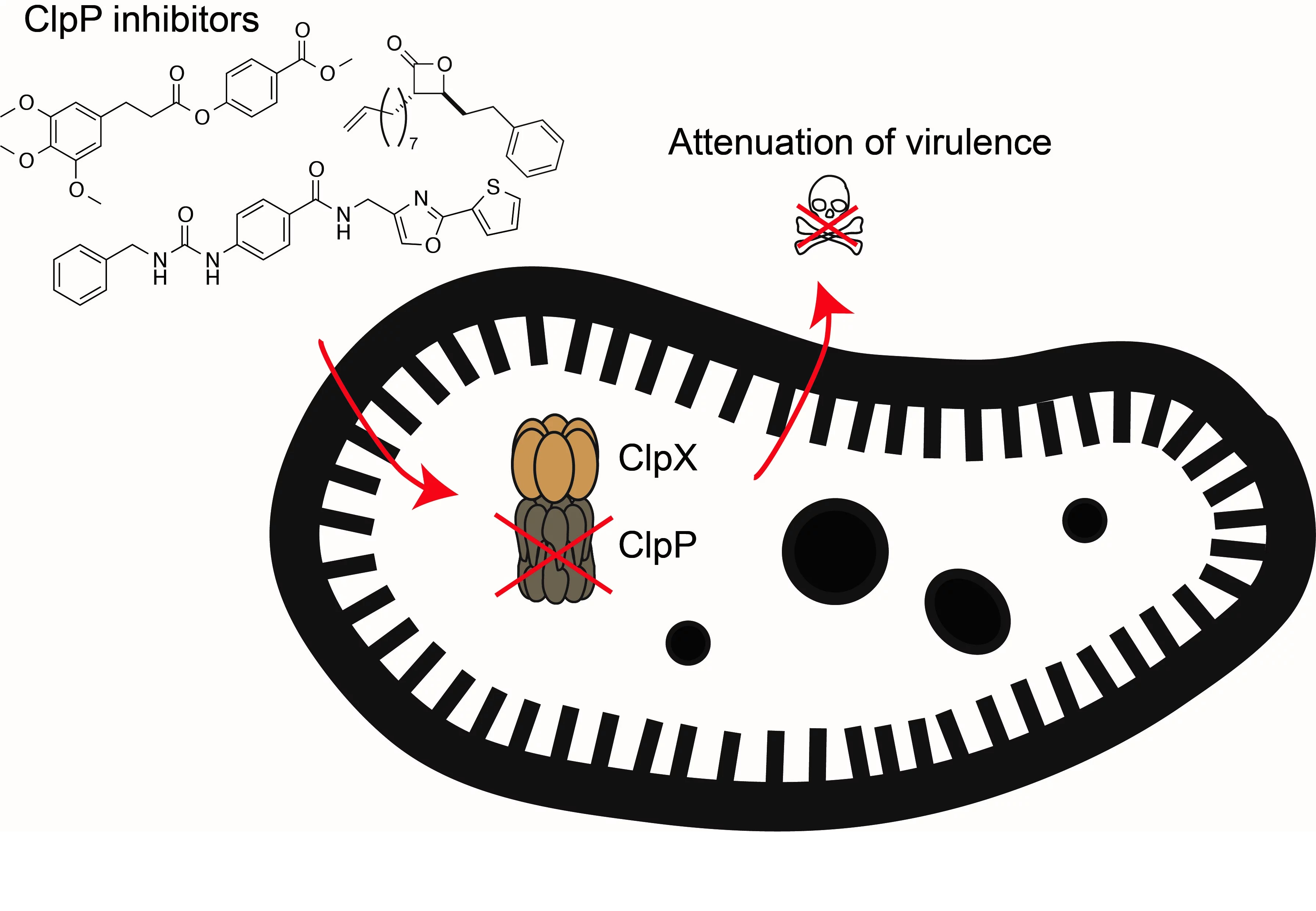
In the search for compounds that block bacterial virulence we focus on a major regulator of toxin production termed caseinolytic protease P (ClpP). Genetic knockouts of ClpP led to the attenuation of S. aureus virulence and we identified the first chemical inhibitor class specific for this enzyme. These inhibitors attenuated virulence of Staphylococcus aureus and multiresistant isolates. In numerous follow-up studies, we have identified novel inhibitor generations, obtained structural information on the binding mode, and unraveled protein dynamics and conformational changes essential to understand the enzyme mechanism of action. In addition, we have also looked for alternative virulence-regulating mechanisms including small molecule-driven sensing of bacteria and the human stress hormone dynorphin.

Moreover, we are interested in the identification of novel antibiotic targets. Small molecules with antibacterial activity, either of natural or synthetic origin, are synthetically equipped with chemical tags to yield probes for target identification via activity-based protein profiling (pioneered by the Cravatt and Bogyo labs) in living bacteria. For example, a recent strategy focussed on the repurposing of existing drugs, i.e. human kinase inhibitors, to kill bacteria. Here we chemically optimized a hit compound of such a screen to yield a potent derivative, termed “PK150”. PK150 is active against methicillin-resistant Staphylococcus aureus (MRSA) clinical isolates, kills difficult-to-treat persister cells, eradicates biofilms, and does not develop resistance in the lab. In depth mode of action studies with a corresponding PK150 probe revealed at least two cellular targets addressed by PK150: overactivation of protein secretion and inhibition of energy metabolism, a so far unprecedented dual mechanism of action. We believe that dysregulation of these targets causes rapid cell death and complicates the development of resistance.
Selected Publications
1. Le, P., Kunold, E., Macsics, R., Rox, K., Jennings, M., Ugur, I., Reinecke, M., Chaves-Moreno, D., Hackl, M.W., Fetzer, C., Mandl, F.A.M., Lehmann, J., Korotkov, V.S., Hacker, S.M., Küster, B., Antes, I., Pieper, D., Rohde, M., Wuest, W.M., Medina, E., Sieber, S.A., "Repurposing human kinase inhibitors to create an antibiotic active against drug-resistant Staphylococcus aureus, persisters and biofilms", Nat. Chem., 2020, 12, 145-158. PMID: 31844194, PMCID: PMC6994260
2. Gatsogiannis, C.°, Balogh, D.°, Merino, F., Sieber, S.A.*, Raunser, S.*, "Cryo-EM structure of the ClpXP protein degradation machinery", Nat. Struct. Mol. Biol., 2019, 26, 946-954. PMID: 31582852, PMCID: PMC6783313
3. Fetzer, C., Korotkov, V.S., Thänert, R., Lee, K.M., Neuenschwander, M., von Kries, J.P., Medina, E., Sieber, S.A., "A Chemical Disruptor of the ClpX Chaperone Complex Attenuates the Virulence of Multidrug-Resistant Staphylococcus aureus", Angew. Chem. Int. Ed., 2017, 56, 15746-15750. PMID: 28906057
4. Gersch, M., Famulla, K., Dahmen M., Göbl, C., Malik, I., Richter, K., Korotkov, V. S., Sass, P., Rübsamen-Schaeff, H., Madl, T., Brötz-Oesterhelt*, H., Sieber, S.A.*, "AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control", Nat. Comm., 2015, 6, 6320, doi:10.1038/ncomms7320. PMID: 25695750