Docking of Biological Macromolecules
Basically all biological processes involve the interaction and complex formation of biomolecules. A full understanding of these interactions requires the three-dimensional (3D) structure of interacting complexes. Experimental structure determination is only possible for a fraction of all possible complexes and may not be feasible for all low-affinity or transient interactions. Therefore, the realistic prediction of protein-protein complexes and protein-nucleic acid complexes is of great importance. We have developed a powerful docking methodology (ATTRACT) based on a coarse-grained representation of partner molecules. The method allows the efficient inclusion of certain types of conformational changes during the computational docking process.
ATTRACT protocols have been developed for protein-protein, protein – nucleic acids and protein-peptides docking, as well as for flexible refinement and fragment-based docking. It also support inclusion of experimental data such as cryo-EM, SAXS, contact and interface data.
Resent new developements
pepATTRACT peptide-protein docking
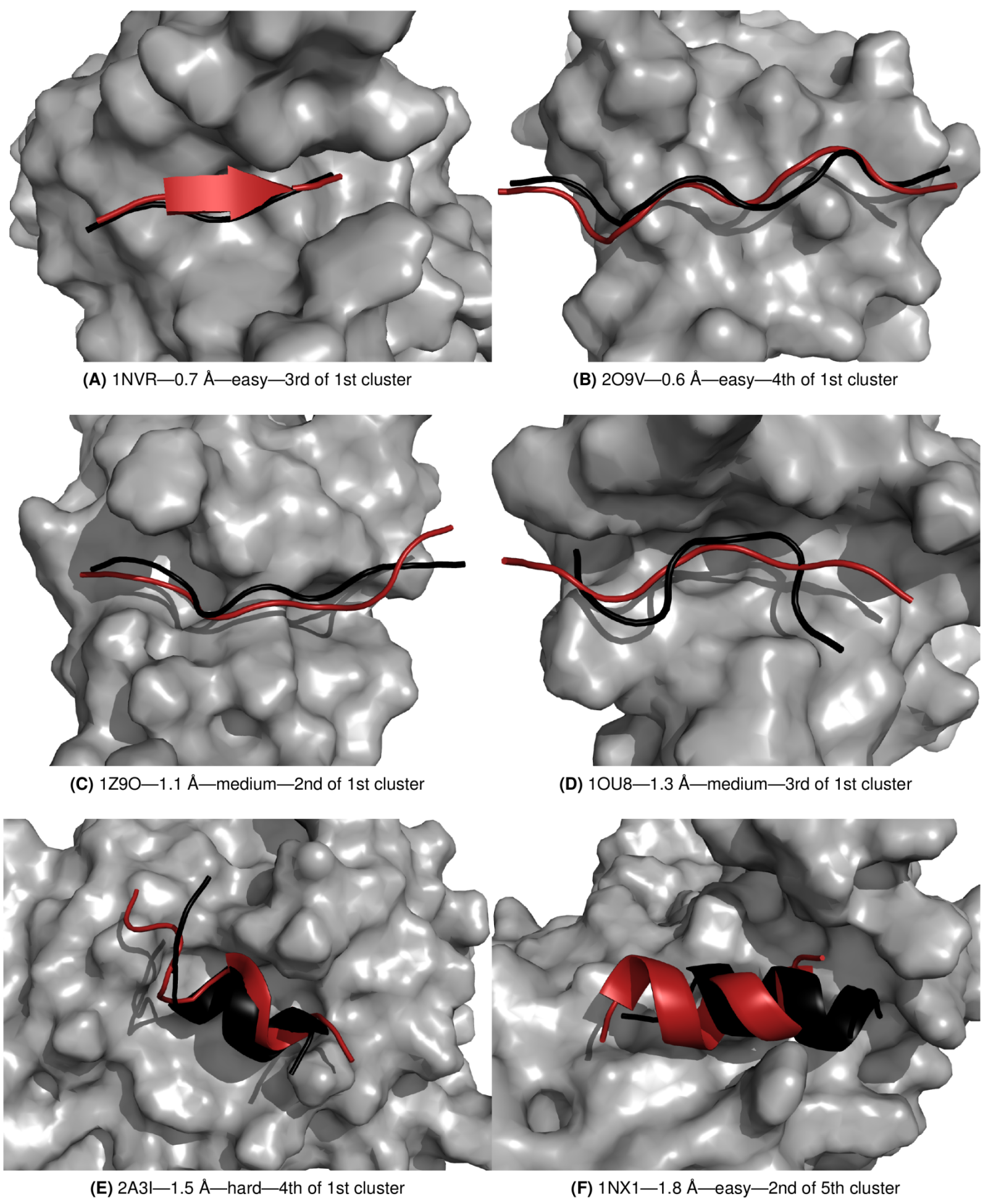
Peptidic interactions make up to 40% of all protein-protein interactions and are involved in many important biological pathways like signal transduction and transcription. Modeling peptide-protein complexes is a highly challenging problem due to the small size of the interface and the high intrinsic flexibility of the peptide. pepATTRACT is one of the first fully blind peptide-protein docking protocols which simultaneously predicts the structure of peptide-protein complexes from the unbound structure of the protein and the peptide sequence. Hence, it requires neither prior knowledge of the binding site nor of the bound peptide conformation. pepATTRACT was tested on a benchmark of 80 known peptide-protein complexes and yielded an overall success rate of 70%. Docking scripts for pepATTRACT can be easily created with the pepATTRACT web-interface.
Rigid-body docking of Biological Macromolecules
Fragment-based docking
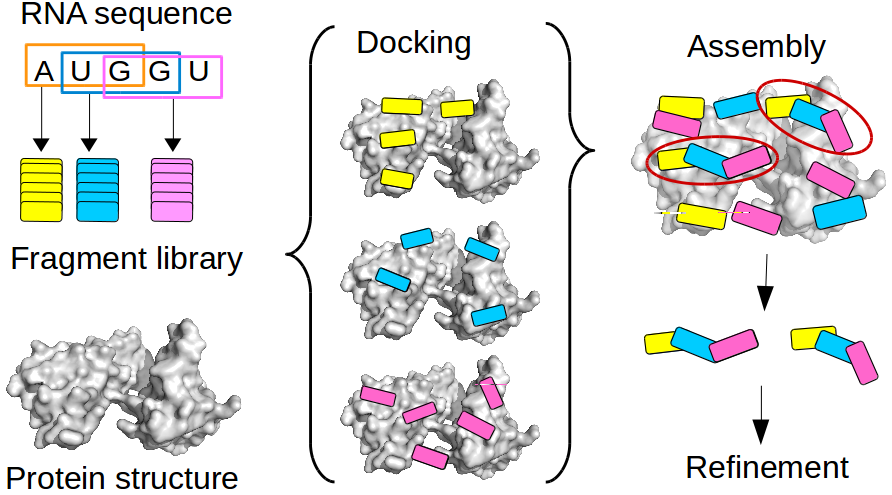
Single-stranded RNA (ssRNA) are highly flexible and can adopt many conformations. Due to the high number of degrees of freedom (DOF) for the conformations of a nucleotide, sampling all possible conformations of e.g. a penta-nucleotide prior to or during is not feasible. To avoid the combinatorial explosion due to accumulation of DOF, we developed a fragment-based approach. This approach allow to model a protein-bound ssRNA based on the structure of the protein and the sequence of the RNA, without any prior knowledge of the RNA binding site or the RNA structure. The conformational diversity of each fragment is sampled by an exhaustive RNA fragment library that we extracted from all the existing experimental structures of protein-RNA complexes. Tested on 9 ssRNA-protein complexes of known structure, the method could model the bound RNA (5 to 11 nucleotide) with a 1.2 to 3 A precision. This is the first time a bound ssRNA could be modeled from sequence with high precision.
Flexible Docking of Biological Macromolecules
iATTRACT flexible interface refinement
We have developed a flexible interface docking refinement, interface-ATTRACT (iATTRACT) for optimizing rigid body docking solutions. The iATTRACT method combines simultaneous full atomistic interface flexibility and rigid body optimizations during docking energy minimization. iATTRACT selects the initial contacts in the docking model on-the-fly and treats these residues as flexible during the refinement. The approach was systematically evaluated on a large benchmark, starting from an enriched decoy set of rigidly docked protein–protein complexes. Large improvements in sampling were observed yielding structures with closer agreement to the native complex for a range of initial deviations. iATTRACT especially optimized the packing of the interface yielding increases in the fraction of native contacts of up to 70%. iATTRACT refinement is available through the ATTRACT protein-protein and peptide-protein docking web-interfaces.