Research Prof. Dr. Michael Groll
Methodology
Primary focus of our research is a combination of biological, chemical, structural and analytical methods to elucidate principles in cellular biology and protein biochemistry, including organic synthesis of new ligands with promise as leads for new medicines. The multi-disciplinary approach of the projects includes techniques such as (A) yeast and human cell culture growth, (B) protein purification, (C) crystallization, (D) diffraction data collection, (E) data evaluation, (F) model building and refinement, (G) structure guided drug design, (H) chemical synthesis, (I) structural and functional analysis of identified ligands in complex with the respective enzymes.

Organic synthesis
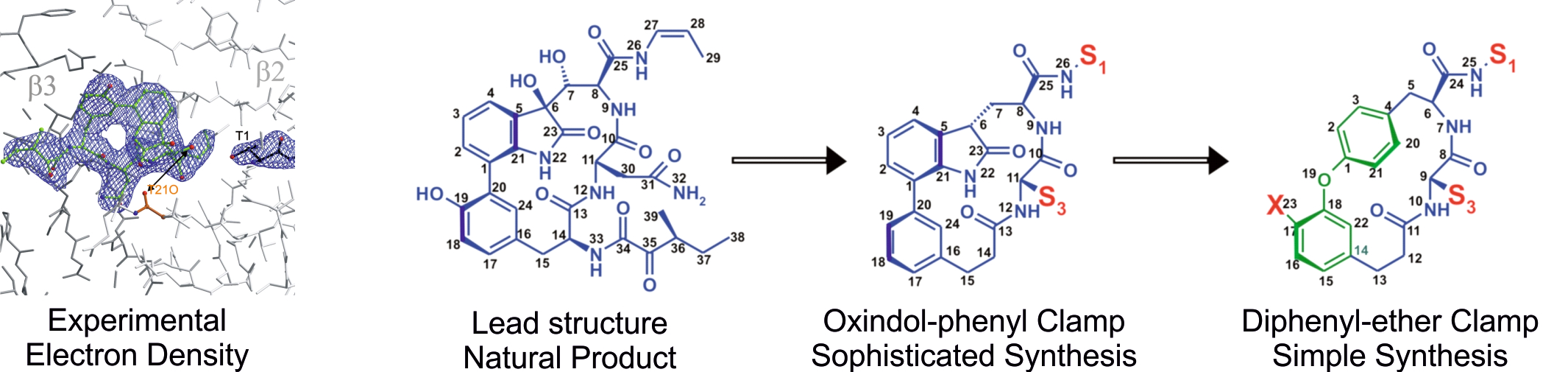
Our organic chemistry lab primarily focuses on the chemical synthesis of novel inhibitors, guided by protein:ligand co-crystal structures and in vitro activity tests. Highly frequent collection of crystal structure data provides rapid feedback for synthetic inhibitor refinement. Our prime example is the eukaryotic 20S proteasome. Along with the improvement of already existent ligands for proteasome subtype selectivity, our efforts also include fragment-based design of novel lead compounds. The aim is to move away from the peptide and warhead paradigm of traditional inhibitor design towards more sophisticated architectures to target novel binding sites. Additionally, synthesis of tailored enzyme substrates helps to develop specialized activity assays or discovering enzymatic reaction trajectories. Applied techniques encompass “standard” organic synthesis, solution/solid phase peptide synthesis, analytic/preparative HPLC/MS, NMR, in vitro/vivo activity assays, X-ray crystallography and automated flash column chromatography. Available techniques for the identification, isolation and characterization of synthetic and natural protein ligands will include. (A) Growth of organisms under varying conditions, (B) Isolation of secretions / natural extracts, (C) NMR screening assay, (D) HTS robotic system, (E) Fluorogenic assay, (F) SSAP assays, (G) Large-scale purification of identified compounds, (H) 2D-NMR analysis for structure identification, (I) crystallization of ligands in complex with the proteasome, (J) structure elucidation, (K) cell culture and activity assays.
Functional and structural characterisation of multifunctional protein complexes
Biological questions often address the understanding of the correlation of molecular form and function. In this context, protein X-ray crystallography gives answers to two major challenges in modern life sciences. Since the method allows the accurate determination of structural details at atomic resolution even for large molecular assemblies, the behavior of entire biological systems can be unraveled. Moreover, novel therapeutic agents may be identified through analysis of drug-target interactions as part of a comprehensive structure guided drug design procedure. In particular, the dissection of the relation between structure and function is a powerful tool for the elucidation of catalytic mechanisms of enzymes, binding modes of interaction partners, the dynamics of a protein and the self-assembly of oligomeric complexes.
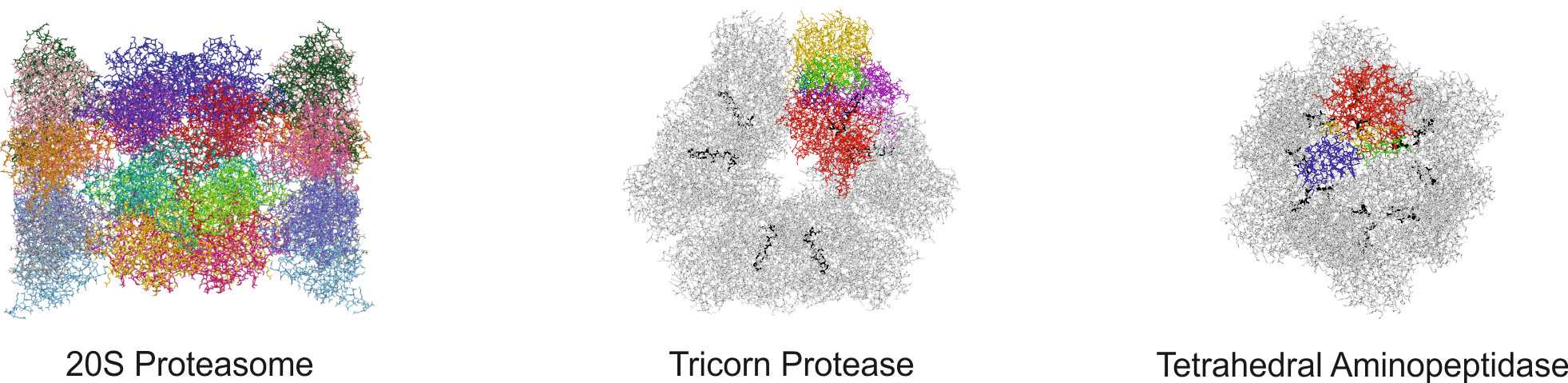
Assembly and regulation of multifunctional protein complexes
The heart of the ubiquitin-mediated degradation pathway, the 26S proteasome, endoproteolytically cleaves most intracellular proteins, thereby maintaining biological homeostasis and regulating many crucial processes in the cell. This hydrolyzing machine comprises more than 30 different subunits, which perform different functions including the recognition, unfolding, translocating and cleavage of protein substrates. Thus, careful assemblage and regulation of the 26S proteasome is essential to ensure correct positioning and function of each subunit, thereby preserving the delicate cellular balance between protein synthesis and degradation.
Development and practical application of protein ligands as innovative drugs
Cancer is the No. 2 cause of death in the western world and one of the most expensive diseases to treat. In addition to targeting kinases in the treatment of cancer, a new approach was achieved to block the 20S proteasome (core particle; CP) activity with the approval for a dipeptide boronic acid analogue, Velcade® (Bortezomib) in May 2003 to be used in the treatment of multiple myeloma patients. Owing to its highly reactive boronic acid head group bortezomib requires intravenous application and is associated with severe side effects such as neuropathy and nerve degeneration. The FDA approval of the second-generation proteasome inhibitor carfilzomib (Kyprolis) for multiple myeloma and mantle cell lymphoma in 2012 once again confirmed the CP as a smart alternative for addressing oncological diseases. Currently, several optimized second-generation proteasome inhibitors are being explored as anticancer drugs in clinical trials, and most of them target both constitutive proteasomes (cCPs) and immunoproteasomes (iCPs). However, selective inhibition of the iCPs, a distinct class of proteasomes predominantly expressed in immune cells, appears to be a promising therapeutic rationale for the treatment of autoimmune disorders including diabetes mellitus type II and multiple sclerosis. Although a few selective agents have already been identified, the determined crystal structure of the iCP will further promote the development and optimization of iCP-selective compounds.
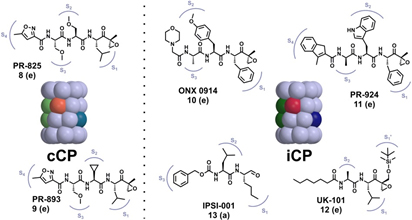
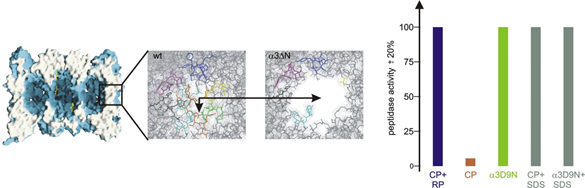
Molecular flexibility and its biological significance
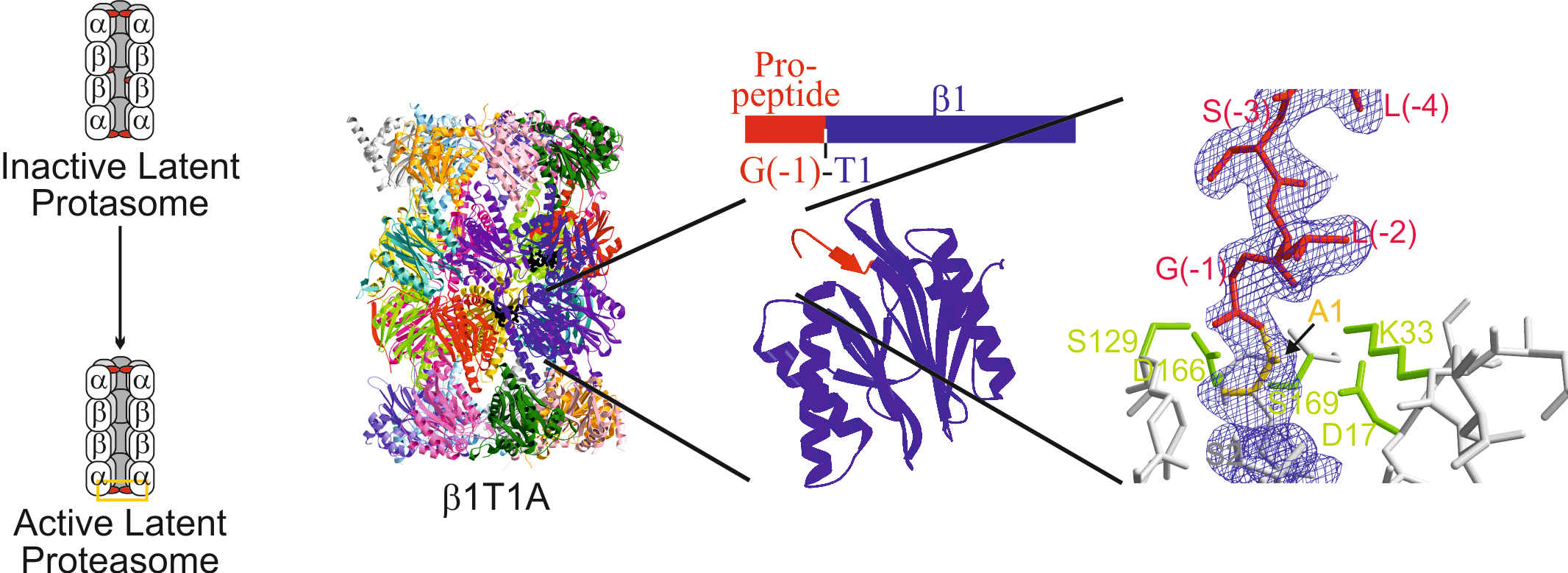
All ATP-dependent proteases of known structure have their proteolytic active sites sequestered within an internal chamber. Protein substrates of these proteases are thought to be unfolded in an ATP-dependent process, before being translocated into the proteolytic chamber. The core particle (CP) of the yeast proteasome is composed of four heptameric rings of subunits arranged in a hollow, barrel-like structure. It is autoinhibited by the N-terminal tails of the outer (α) ring subunits. Crystallographic analyses show that deletion of the tail of the α3-subunit opens a channel into the proteolytically active interior chamber of the CP, thus derepressing peptide hydrolysis. In the latent state of the particle, the tails prevent substrate entry by imposing topological closure on the CP. Inhibition by the α-subunit tails is relieved upon binding of the regulatory particle to the CP to form the proteasome holoenzyme.
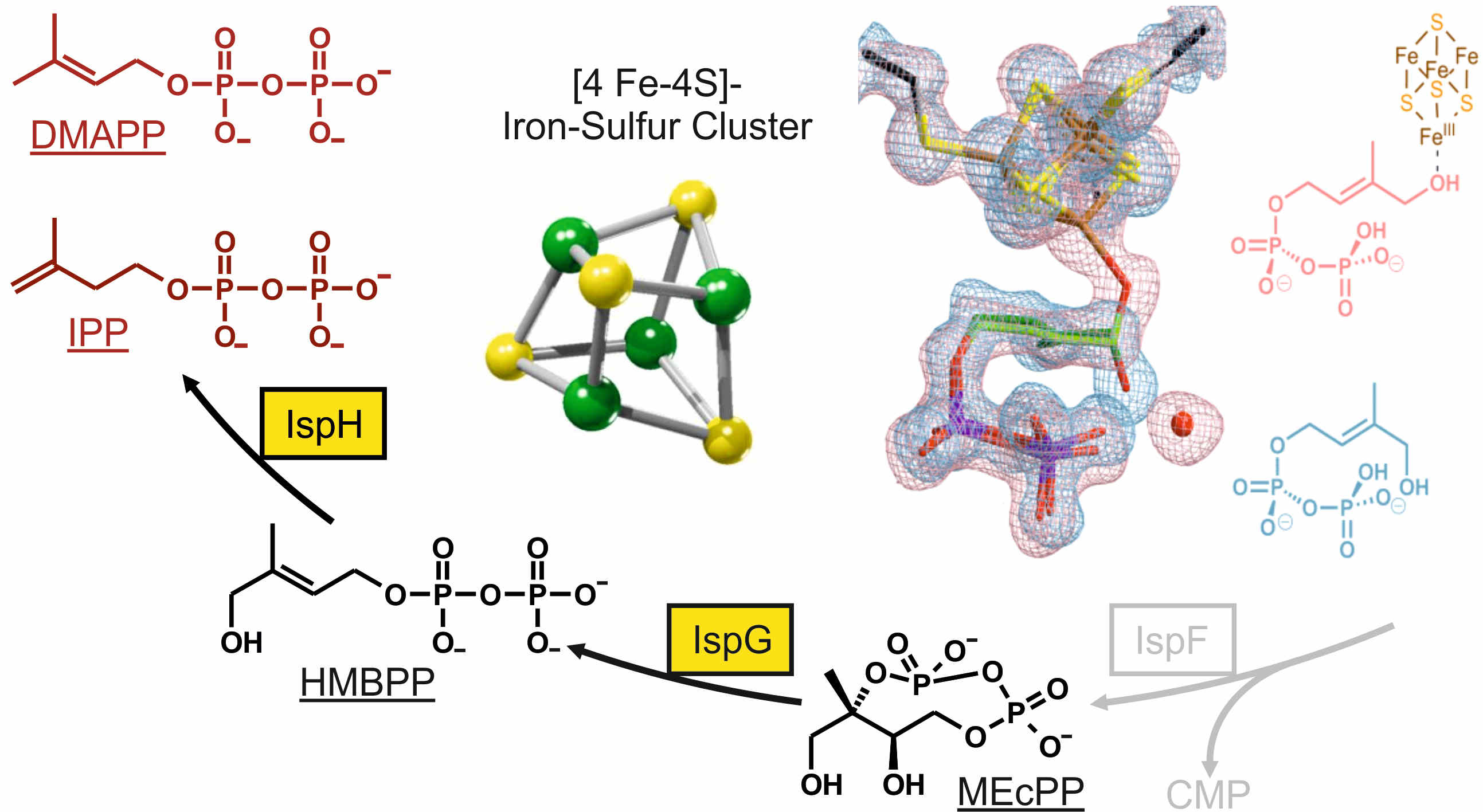
Molecular targets for drug development
The non-mevalonate pathway of terpenoid biosynthesis is essential in many eubacteria including the major human pathogens Mycobacterium tuberculosis and the protozoa Plasmodium spp. as well as the plastids in plants. The metabolic route is absent in mammals and is therefore qualified as a promising target for new anti-infective drugs and herbicides. Biochemical and structural knowledge about all enzymes involved in the pathway provide the basis for the discovery and the development of inhibitors by high-throughput screening of compound libraries and/or structure-based rational design. Particularly, our work is focused on the structural and functional studies on the iron-sulfur-proteins IspG and IspH, which might open up new routes to inhibitor design against diseases such as tuberculosis and malaria.
Identification and characterization of natural enzymatic modulators
The 20S proteasome is a conserved target that is addressed by various pathogenic bacteria and fungi during their pathogenic life-stages. The second generation of proteasome inhibitors, which exhibit far enhanced properties in terms of off-target reactivity and adverse clinical side effects, use the lead motives of these highly specific and optimized secondary metabolites. However, the potency of proteasome modulating agents with respect to many other applications fuels the search for more compounds that could serve as template structures for further drug development. Our current aim is the identification and characterization of such natural products. Therefore, we use diverse techniques like UV-VIS and fluorescence high-throughput screenings as well as NMR, gel-shift and mass spectrometry assays. Once a hit is identified, we structurally characterize the compound by the means of 2D-NMR and x-ray analysis. Finally, its in vivo activity is evaluated in our cell culture facility by cell viability and pathway specific assays.
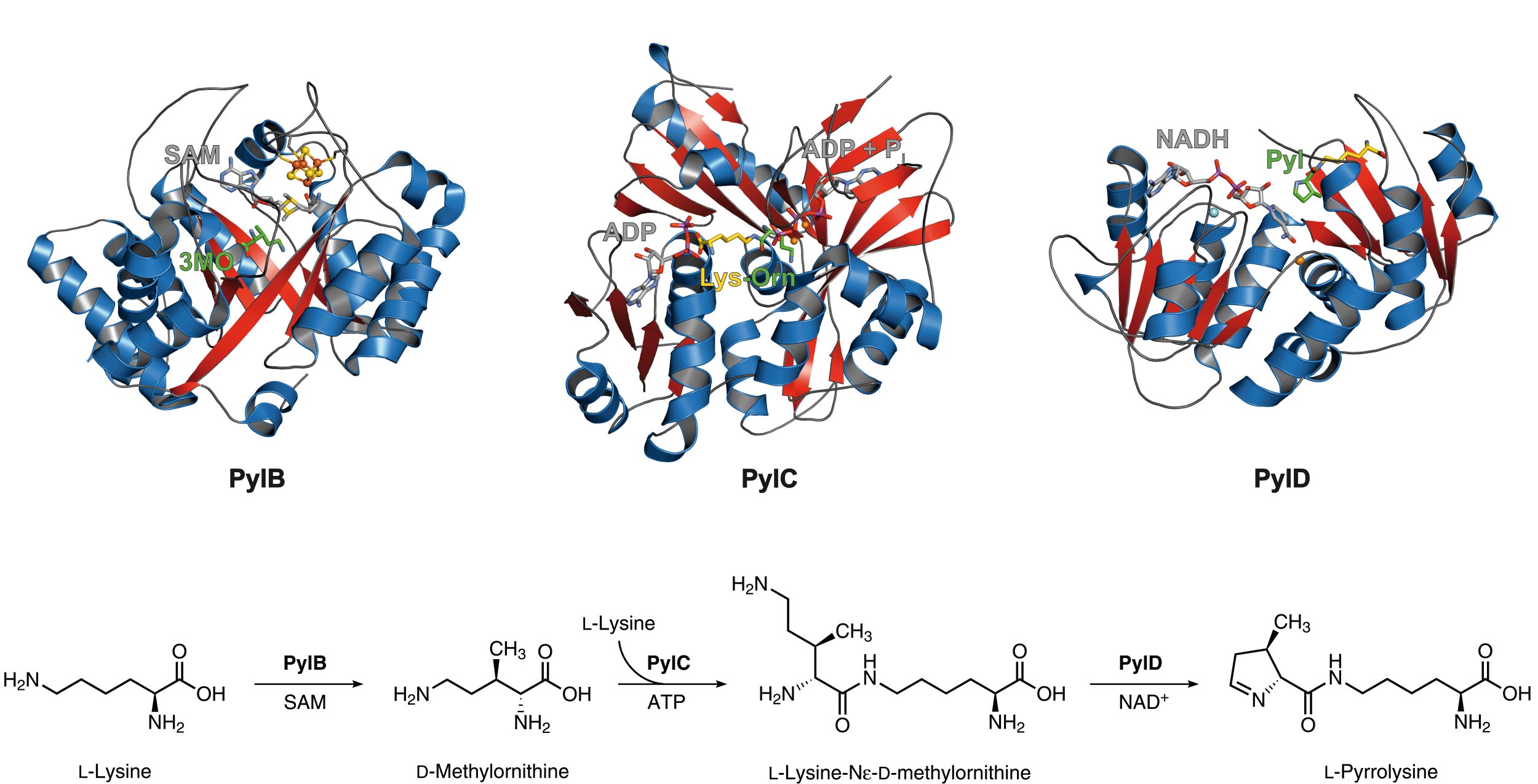
Biosynthesis of the amino acid pyrrolysine
Pyrrolysine is the 22nd genetically encoded amino acid and is incorporated via the read-through of a so-called ‘amber’ stop codon (UAG), which normally terminates the translation. The unusual amino acid is made by the consecutive action of three proteins specified by PylB, PylC and PylD. PylB, which harbors an [4Fe-4S] iron-sulfur cluster and S-adenosylmethionine as cofactor, converts L-lysine by a radical fragmentation-recombination mechanism to 3R-methyl-D-ornithine that is ATP-dependently linked to the ε-amino group of a second lysine molecule by PylC. Interestingly, PylC possesses a second ATP binding site which is not essential for catalysis. Mutagenesis studies reveal that the additional adenine nucleotide is responsible for protein stability and substrate coordination. Finally, the PylC product is oxidized by the PylD coenzyme NAD+, followed by a ring-closure, resulting in the final product pyrrolysine. Complex structures of PylD with the substrate or product revealed an induced-fit mechanism upon substrate binding where the N-terminal helix breaks and encloses the active site cavity.
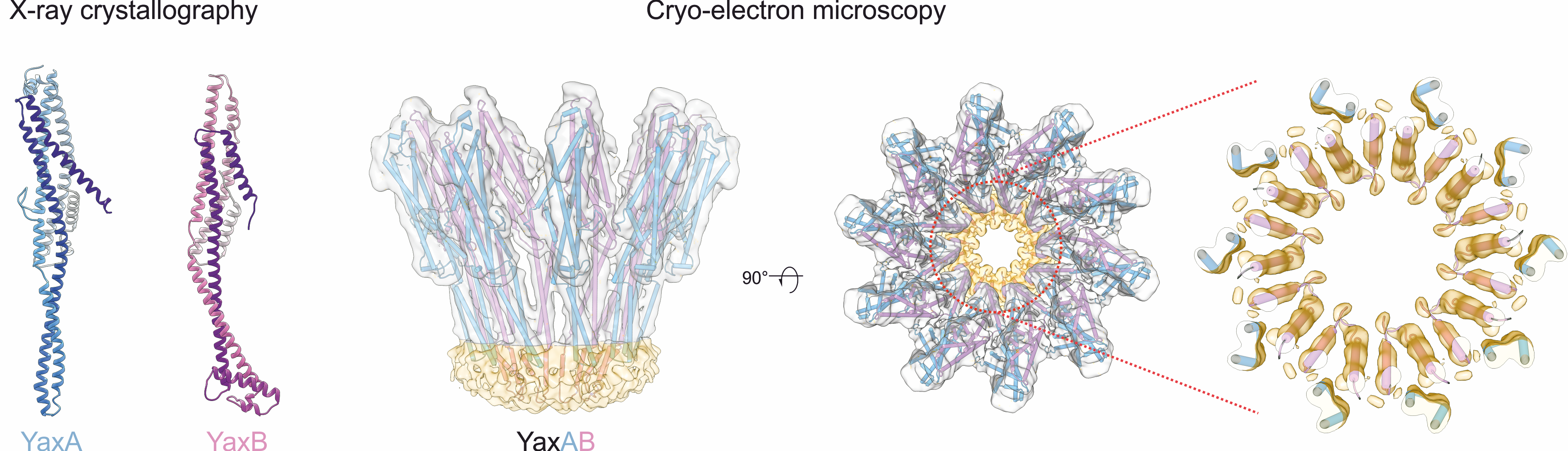
Structure and mechanism of pore-forming toxins
Pore-forming toxins that transform from soluble to membrane-bound states are found ubiquitously throughout prokaryotes and eukaryotes. The YaxAB system from the human pathogen Yersinia enterocolitica is a putative virulence factor with orthologous toxins identified in other mammalian, insect and plant pathogens. To unravel the structural basis for its two-component assembly mechanism, we applied a multi-disciplinary approach combining crystallography (with toxin components YaxA and YaxB), cryo-electron microscopy (visualizing the reconstituted pore) and biochemical assays (to dissect functional roles of each subunit in pore formation). Together, our data point to a sequential mode of toxin formation, whereby YaxA and YaxB consecutively assemble the physiological complex in host membranes. These mechanistic insights might serve to target YaxAB and its orthologues for therapeutic and biotechnological applications in the future.