New Publication in the Journal of Cheminformatics
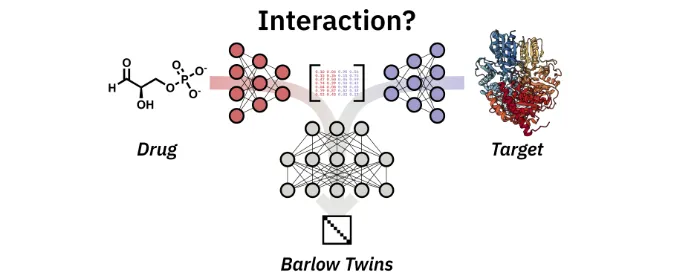
Accurate prediction of drug–target interactions is critical for advancing drug discovery. By reducingtime and cost, machine learning and deep learning can accelerate this laborious discovery process. In a novelapproach, BarlowDTI, we utilise the powerful Barlow Twins architecture for feature-extraction while consideringthe structure of the target protein. Our method achieves state-of-the-art predictive performance against mul-tiple established benchmarks using only one-dimensional input. The use of our hybrid approach of deeplearning and gradient boosting machine as the underlying predictor ensures fast and efficient predictions with-out the need for substantial computational resources. We also propose the use of an influence method to investi-gate how the model reaches its decision based on individual training samples. By comparing co-crystal structures, we find that BarlowDTI effectively exploits catalytically active and stabilising residues, highlighting the model’s ability to generalise from one-dimensional input data. In addition, we further benchmark new baselines against existing methods. Together, these innovations improve the efficiency and effectiveness of drug–targetinteractions predictions, providing robust tools for accelerating drug development and deepening the under-standing of molecular interactions. Therefore, we provide an easy-to-use web interface that can be freely accessedat https://www.bio.nat.tum.de/oc2/barlowdti.
Schuh, M. G., Boldini, D., Bohne, A. I. & Sieber, S. A. “Barlow Twins deep neural network for advanced 1D drug–target interaction prediction”, J. Cheminform.
Link: https://doi.org/10.1186/s13321-025-00952-2
This reprint is licensed under CC-BY 4.0